Professional Chemical Supplier
Elements leading to a rational design of cyclodextrin-based formulations
Elements leading to a rational design of cyclodextrin-based formulations
Cyclodextrins are a frequently used excipient in the pharmaceutical industry, employed in multiple dosage forms and with multiple purposes simply because of their numerous positive properties, including being generally well tolerated and safe. In this short article, René Holm, Professor in pharmaceutical physical chemistry at the University of Southern Denmark, provides a few examples of how a rational formulation design for a cyclodextrin solution can be defined for some important parameters.
Cyclodextrins (CDs) principally belong to the field of supramolecular chemistry.1 In their pharmaceutical application they are used as molecular containers (hosts) that can form inclusion complexes with drug compounds, according to the equilibrium for a 1:1 system:
CD + compound ⇔ CD-compound (1)
which can be expressed quantitatively by the stability constant, Kcomplex:
Kcomplex =
[CD – compound]
___________________ (2)
[CD]∙[compound]
CDs are cyclic oligosaccharides, produced from enzymatic degradation of starch. They consist of 1,4-linked glucopyranose units positioned in a truncated conelike structure.2 The inside of the cone is referred to as the CD cavity and it is relatively hydrophobic, lined with hydrogen atoms and glycosidic oxygen bridges, causing a high electron density within the cavity.1 The exterior of the CD is covered by hydroxyl groups providing hydrogen bond donors and acceptors, making the surface hydrophilic.3 The size of the CD cavity is determined by the number of glucopyranose units, with the most common CDs containing six to eight and referred to as α-, β and γ-CD, respectively.4,5 The three common CDs have different chemical and physical properties, eg, molecular weight, cavity size and aqueous solubility.4
The chemical structure of CDs can be modified by substitution at the hydroxyl groups, thus creating derivatives of CDs.2 Modified CDs have higher solubility and even substitution with hydrophobic side groups leads to improved solubility.2 Today numerous functionalised CDs exist; the most common modifications are the methyl-, hydroxypropyl- and sulfobutylether modifications.6 CDs have been used in formulations administered via multiple routes including oral, subcutaneous, intravenous and ocular. They are most frequently used as a solubilising excipient, but can also be used for other purposes, including chemical stabilisation and taste masking.
The chemical structure of CDs can be modified by substitution at the hydroxyl groups
When used in a formulation it is important to understand what is critical relative to the purpose of the addition of the CD. For example, if the purpose is to use the CD as a chemical stabiliser, ie, to prevent degradation of the compound, then the highest benefit is obtained in a system where the fraction of compound bound within the CD is highest. This must be considered in the formulation design, with a design that will not necessarily maximise the fraction that can be solubilised, requiring a different composition of the formulation. Understanding the equilibriums in CD systems is therefore key to make an optimal formulation. Fortunately, CD formulations generally behave according to the learnings from classical physical pharmacy, hence a rational design can be defined by investigations of the relevant equilibriums. In this article, some of these elements will be presented to provide a flavour of the approach that could be chosen, with a focus on liquid formulations.
Selecting the cyclodextrin type
As describe above, there are three main types of CDs, as well as substituted versions of these – thus, the right one must be defined. This is often achieved through relatively simple phase solubility studies, ie, solubility studies with increasing amounts of CDs at a defined pH. Based upon these experiments, a CD can normally be identified that provides the highest apparent solubility. This complex then needs some additional characterisations to make a truly rational development.
Defining the pH, buffer and buffer concentration
Many drug compounds have acid/base properties, ie, a pKa value. In general, the charged version of the compound has a significantly lower stability constant than the uncharged compound. It therefore makes sense to investigate the stability constant of the different species, ie, all the different charged and uncharged versions of the drug compound. This can be used at all times to estimate the apparent stability constant at a given pH, as recently demonstrated by Samuelsen et al.7 An example of the equilibrium scheme for interaction between CD and an amphoteric guest molecule is provided in Figure 1.
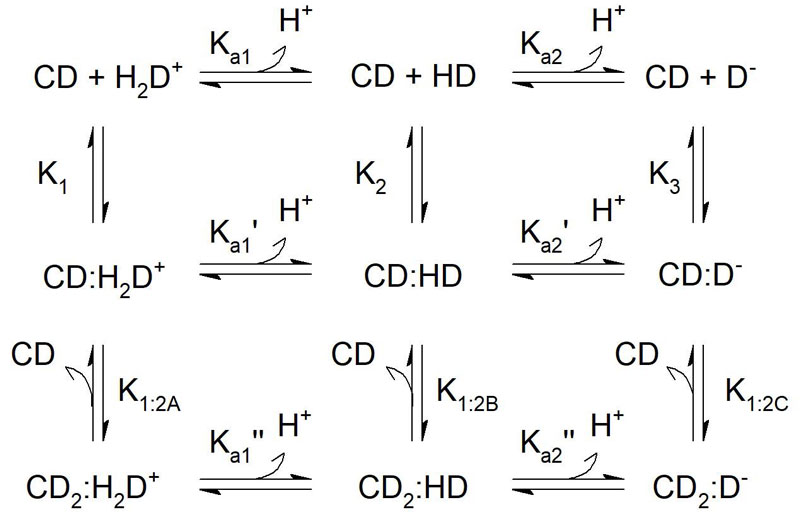
Figure 1: Equilibrium scheme of possible interactions between CD and ionisable guest.
This example considers 1:1 and 1:2 binding mechanism to an amphoteric guest molecule.
If possible, these investigations should be carried out by adjusting the pH with the addition of small strong acid/bases as organic buffer can interact with the CD.8 One of the strongest techniques to use for these investigations is isothermal titration calorimetry (ITC), as it provides the scientist with all thermodynamic parameters of the interaction in a single experiment. However, other techniques could also be considered. In addition, investigating where on the drug molecule the complexation occurs may be beneficial, eg, by carbon-13 (13C) nuclear magnetic resonance (NMR) or rotating‑frame nuclear Overhauser effect spectroscopy (ROESY) NMR. These activities are not critical, but they provide a good understanding of the chemical space.
Selection of the solution’s final pH is a combination of multiple factors: the administration site, optimal stability of the drug product and optimal solubility. When a pH range is defined, a buffer is normally used to stabilise the pH in this range – if possible, this buffer should be selected to have no interaction with the CD, as this would form a competitive complexation. Interaction of the buffer with the CD would lead to a lower apparent stability constant for the drug substance and thereby decrease the solubilising effect of the added CD.7 Also, the buffer concentration should be chosen at the lowest possible range for the same reason.
Formulation robustness and manufacturing
If a substituted CD is selected, several additional sensitivity investigations would be recommended, in addition to examining the effect of pH.
For substituted CDs this would include investigating the influence of the degree of substitution on the stability constant, as there may be batch-to-batch variations in the substitutions. Thus, clarification of the sensitivity is recommended.
Another important parameter is temperature, as both the solubility of the compound in water and the stability constant may vary as a function of temperature. Often, a decrease in aqueous solubility is observed, whereas the stability constant for the CD complexation increases. This should be confirmed experimentally, but could be simulated theoretically as demonstrated in Figure 2.
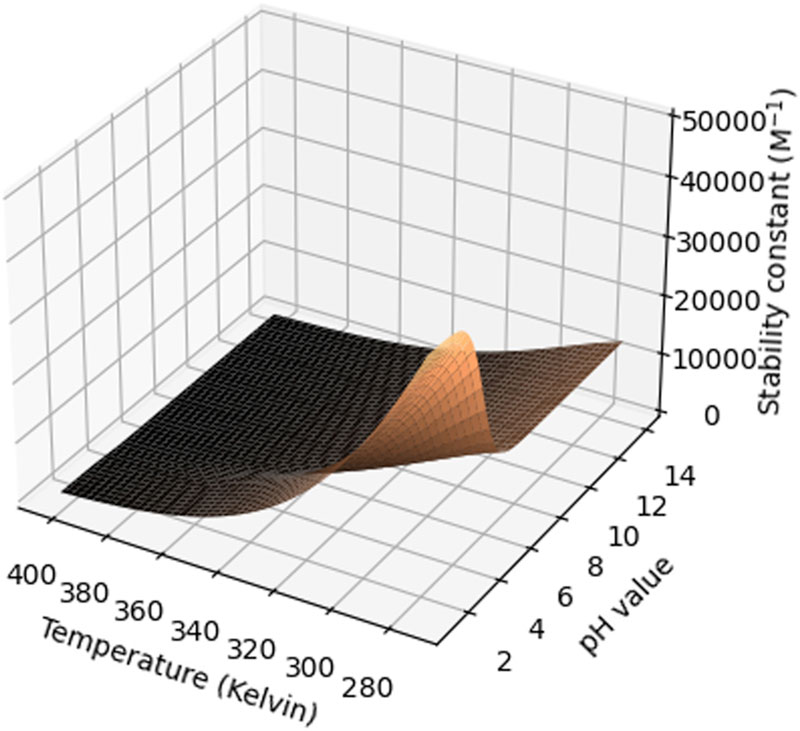
Figure 2: Surface plot of the stability constant (M-1) as a function of temperature (kelvin) and pH for the β-CD:ibuprofen complex (reproduced from Samuelsen et al.7 with permission).
This figure shows a modelling of two factors at the same time, but more could, in principle, be simulated if relevant. It is important to consider that many of the equilibriums in the formulation may be interconnected. If the characterisation work has been conducted thoroughly and the relevant stability constants determined, it is possible to generate similar theoretical simulations based upon classical equilibrium theory, which can subsequently be verified experimentally supporting the line of thought in the ICH Q8 (R2) guideline for formulation development.
Key factors to consider when working with CD-based formulations
Manufacturing of CD solutions is, in principle, a relatively simple process. CDs have a good and fast solubility in water, and are sufficiently stable to undergo a classical autoclavation. It is important to be aware when producing solutions that may encounter temperature changes, eg, to increase solubilisation rate or autoclavation, that pKa values can be affected for buffers and compounds with a temperature sensitivity.9 This means the pH-based equilibriums may change during manufacturing – at worst, leading to precipitation of the drug compound during the process, which could be simulated if the stability constants for the relevant equilibriums have been determined. Most drug products contain relatively low amounts of the active compound, so this process is often defined by the buffer and is why the buffer should be selected with this consideration in mind. Though not an exhaustive list, this article provides a flavour of how basic physical chemical insights can be used in the fundamental development work of CD‑based formulations.
About the author
Dr René Holm is a professor in pharmaceutical physical chemistry at the University of Southern Denmark, a position he has had since April 2021. Before that, he worked for more than 20 years in the pharmaceutical industry. Professor Holm is a (co)‑author and (co)-inventor of more than 230 peer-reviewed papers, book chapters and patents in the field of pharmaceutical sciences. Professor Holm researches in the fields of cyclodextrins and the physical chemical understanding of interactions with compounds and pharmaceutical excipients as well as long-acting injectables for different modalities, with focus on the science behind the formulation systems.